


COVID-19 CEPIVA SO EKSPERIMENTALNA CEPIVA
*
Poslanec v Evropskem parlamentu: zaustavite ta eksperiment na ljudeh
*
cepiva so eksperimentalna- odgovor JAZMP
*
Pri poslušanju radijske oddaje RTV SLO dne 27.maja 2021 z naslovom Cepiva proti covidu niso eksperimentalna, smo odkrili napačno izjavo kliničnega mikrobiologa dr. Alojza Ihana, da COVID-19 cepiva niso eksperimentalna cepiva.
*

*
Dr. Ihan odgovarja na vprašanje voditelja, zakaj je Evropska agencija za zdravila (EMA) izdala pogojno dovoljenje za COVID-19 cepiva in ali so v eksperimentalni fazi. Odgovor dr. Ihana: Ne, niso v eksperimentalni fazi...pogojno dovoljenje ni status kliničnega eksperimenta. Ti eksperimenti so opravljeni, ko se konča 3. faza kliničnih testiranj, pogojno pa je zaradi tega, ker se 3. faza običajno vleče nekako do drugega leta po začetku testiranja. To je običajno pri cepivih, čeprav vemo da se vse pomembne komplikacije oz. neželeni učinki pojavijo v prvih mesecih po cepljenju, vendarle dve leti traja ta postopek običajno.
Seveda je pa možno v primeru potreb, ko se presodi da bi bila škoda tega čakanja bistveno večja kot če se dobi dovoljenje prej, je možno dajati začasno dovoljenje zaradi presoje, da je to nujno. S tem lahko prihranimo veliko človeških življenj in zato so ta cepiva dobila pogojno dovoljenje. To pomeni, da bo doba opazovanja še vedno trajala dve leti, vendar so regulatorni organi presodili, da bi bila cena, ki bi jo plačali za dvoletno čakanje zanesljivo mnogo mnogo večja, kar se tiče cene človeških življenj, kot pa se bo zgodilo to v primeru, ko so cepiva dobila pogojno dovoljenje in lahko delujejo že takoj. In to se je izkazalo kot izjemno pametno, kajti zdaj je cepljenih že več kot milijarda ljudi in države, kjer je precepljenost že visoka, kot je recimo Izrael, so že lahko naredila jasne statistične analize. V teh državah je jasno, da so v bolnišnicah praktično samo ljudje, ki niso bili cepljeni.
Vir: https://radioprvi.rtvslo.si/2021/05/ultrazvok-ihan-covid-cepljenje/ (0d 3.35 do 6.12 min)
*
Covid-19 cepiva imajo pogojno dovoljenje za uporabo ( v sili- emergency use): https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation
*
Po pregledu literature o eksperimentalnih cepivih smo ugotovili:
1. Največji ameriški vir podatkov o zdravilih- drug.com navaja naslednji opis: Moderna cepivo proti COVID-19 je eksperimentalno in vsa njegova tveganja še niso znana. Cepivo covid-19 ne vsebuje koronavirusa in vam ne more dati covid-19.
Vir: https://www.drugs.com/mtm/moderna-covid-19-vaccine.html
*
2. Ameriški Nacionalni inštitut za zdravje dne 12. januarja 2021 navaja: Moderna eksperimentalno cepivo je zelo učinkovito
Vir: https://www.nih.gov/news-events/nih-research-matters/experimental-coronavirus-vaccine-highly-effective
*
3. Kaj meni združenje American Frontline Doctors (AFLDS) z besedo "eksperimentalno cepivo"? Po mnenju ameriške Uprave za hrano in zdravila (FDA), "Preiskovalno zdravilo lahko imenujemo tudi eksperimentalno zdravilo, ki se preučuje, da se ugotovi ali se vaša bolezen ali zdravstveno stanje izboljšuje med jemanjem." Zahtevki Pfizer-ja, Moderne in AstraZenece pravilno prepoznajo svoje nove agente kot "preiskovalne", kar je normalno v tej zelo zgodnji fazi razvoja. Vsi kandidati za COVID19 cepivo so kategorizirani kot eksperimentalni iz naslednjih razlogov:
• farmacevtska podjetja so zaprosila za status uporabe v preiskavi
• neželeni dogodki bodo poravnani v skladu s pravnim standardom za eksperimentalna zdravila
• prejemniki so vpisani kot preiskovanci v zdravniško preskušanje za zbiranje podatkov o neželenih učinkih.
• osebe so vsaj dve leti vključene v sistem za sledenje farmakološkega nadzora (pharmaco-vigilance)
• številne skupine oseb niso bile raziskane, vključno z: predhodnimi bolniki s COVID-19, nosečnicami, mladimi, starejšimi
• ni objavljenih podatkov o študijah na živalih
Vir: https://www.americasfrontlinedoctors.com/wp-content/uploads/Vaccine-PP.pdf
*
4. Uprava za hrano in zdravila (FDA) je Pfizer-ju dovolila testiranje eksperimentalnega COVID-19 cepiva na ameriških otrocih. 16. oktobra 2020 je Pfizerjev predsednik uprave naznanil, da bo družba konec novembra pri FDA za svoje eksperimentalno cepivo proti COVID-19 - BNT162b2 vložila dovoljenje za uporabo v sili.
Vir: https://childrenshealthdefense.org/defender/fda-pfizer-experimental-covid-vaccine-children/
*
5. Večina ljudi se ne zaveda, da je mnogo na novo licenciranih cepiv v kliničnih preskušanjih faze 4. Pri novih cepivih proti COVID-19, ki še niso bila licencirana, so potrošniki brez vedenja preiskovanci v množičnem kliničnem preskušanju.
Popoln varnostni profil cepiva v času razširjene uporabe ni znan. Varnostni profil postane bolj jasen šele po poročilu o poškodbah in smrtih, povezanih s cepivom. Varnostni profil cepiv proti COVIDu se na primer še vedno razvija, saj se kopičijo poškodbe in smrtni primeri.
Zato je ključnega pomena, da potrošniki in zdravstveni delavci poročajo o neželenih dogodkih, povezanih s cepivi. Po študijah pa poročajo le o približno 1 % neželenih dogodkov.
Vir: https://childrenshealthdefense.org/defender/serious-injuries-merck-gardasil-hpv-vaccine-significantly-underreported/
Label your annual report as an “Annual Status Report of Postmarketing Study Requirement/Commitments” and submit it to the FDA each year within 60 calendar days of the anniversary date of this letter until all Requirements and Commitments subject to the reporting requirements under section 506B of the FDCA are released or fulfilled. These required studies are listed below: 1. Deferred pediatric Study C4591001 to evaluate the safety and effectiveness of COMIRNATY in children 12 years through 15 years of age. Final Protocol Submission: October 7, 2020 Study Completion: May 31, 2023 Final Report Submission: October 31, 2023
https://www.fda.gov/media/151710/download
*
6. V skladu z ameriško zvezno zakonodajo delodajalci in univerze ne morejo zakonito določiti cepiv za COVID-19 kot obvezna, ker so to nelicencirani izdelki z dovoljenjem za uporabo v sili (EUA- Emergency Use Authorization status), ki so po definiciji eksperimentalna cepiva.
Obvezna uporaba izdelkov, pooblaščenih za status odobritve za uporabo v sili (EUA), krši ameriško zvezno zakonodajo, kot je podrobno opisano v naslednjih pravnih obvestilih.Vsa CEPIVA PROTI COVIDU-19, COVID PCR in antigenski testi in maske so avtorizirana samo za uporabo v sili (EUA), niso odobrena ali licencirana s strani zvezne vlade. Njihova dolgoročna varnost in učinkovitost ni bila dokazana.
Proizvodi z dovoljenjem za uporabo v sili (EUA) so po definiciji eksperimentalni, zaradi česar je ljudem treba dati pravico, da jih zavrnejo. Po Nürnberškem zakoniku, temelju etične medicine, nihče ne sme biti prisiljen, da sodeluje v medicinskem poskusu. Privolitev posameznika je "absolutno bistvena".
V začetku tega leta sta Mary Holland, predsednica in generalna svetovalka Childern’s Health Defense ter odvetnik Greg Glaser naznanila, da ameriški zvezni zakon delodajalcem prepoveduje, da bi EUA COVID cepiva, EUA COVID-19 teste in maske (z dovoljenjem za uporabo v sili) predpisali kot obvezne.
Pravna ekipa Childern’s Health Defense je napisala tri pravna obvestila, ki jih lahko vsakdo, ki se sooča z obveznim covid cepivom, covid testom ali mandatom maske, uporabi za obveščanje delodajalcev in univerz, da kršijo zvezno zakonodajo:
- Pravni obrazec za eksperimentalne covid maske: https://childrenshealthdefense.org/wp-content/uploads/chd-notice-for-eua-masks.pdf
- Pravni obrazec za eksperimentalne covid teste: https://childrenshealthdefense.org/wp-content/uploads/chd-notice-for-eua-testing.pdf
- Pravni obrazec za eksperimentalna covid cepiva: https://childrenshealthdefense.org/wp-content/uploads/chd-notice-for-eua-vaccines.pdf
Ameriško Zvezno sodišče je razsodilo, da Ameriška vojska ne more predpisati cepiv za uporabo v sili (EUA) kot obvezna za vojake. Doe #1 v. Rumsfeld, 297 F.Supp.2d 119 (2003). Sodišče je razsodilo: "... Združene države ne morejo zahtevati, da pripadniki oboroženih sil služijo kot poskusni prašički za eksperimentalna zdravila." Nobeno sodišče še ni potrdilo obveznosti za cepivo z dovoljenjem za uporabo v sili (EUA).
Vsak subjekt ali organizacija, ki zahteva obvezno cepljenje COVID-19, testiranje za COVID-19 ali maske, ki imajo dovoljenje za uporabo v sili, krši zvezno zakonodajo in se bo lahko soočila s tožbami, če ne bodo dovolili izjem ali alternativ."
Vir: https://childrenshealthdefense.org/defender/resources-federal-law-prohibits-mandates-emergency-use-covid-vaccines-tests-masks/
*
7. Aplikacija za preiskovalno novo zdravilo IND (op. prev.: preiskovalno zdravilo je eksperimentalno zdravilo)
Dovoljenje za uporabo v sili (Emergency Use) za preiskovalno novo zdravilo (IND) omogoča Upravi za varno hrano in zdravila, da odobri uporabo eksperimentalnega zdravila v izrednih razmerah, ki ne omogoča časa za predložitev aplikacije za preiskovalno novo zdravilo (IND) v skladu z 21CFR , Sec. 312.23 ali Sec. 312.20. Uporablja se tudi za bolnike, ki ne izpolnjujejo meril obstoječega protokola študije ali če odobreni protokol študije ne obstaja.
Vir: https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application#Introduction
*
8. Rusija in Kitajska sta na podlagi razmeroma nizkega števila podatkov sprožila množično uporabo domačih cepiv, ki jih sponzorira država. Takšne skrajšane regulativne poti in hitre uvedbe, ki se še vedno pogosto štejejo za eksperimentalne posege v okviru izrednega stanja na področju javnega zdravja v mednarodnem smislu, so brez primere.
Vir: https://www.thelancet.com/journals/laninf/article/PIIS1473-3099(20)30923-3/fulltext
*
9. Eksperimentalno zdravilo je zdravilo ali cepivo, ki še ni prejelo odobritve vladnih regulativnih organov za rutinsko uporabo v humani ali veterinarski medicini, čeprav lahko ozdravi virus/bolezen.
V Združenih državah Amerike je organ, odgovoren za odobritev, ameriška Uprava za hrano in zdravila (FDA), ki mora pred testiranjem v kliničnih preskušanjih na ljudeh odobriti status snovi kot preiskovalno novo zdravilo (Investigational New Drug-IND). Status preiskovalnega novega zdravila (IND) od sponzorja zdravila zahteva, da predloži vlogo za IND, ki vključuje podatke iz laboratorijskih in živalskih testov za varnost in učinkovitost zdravila.
Vir: https://en.wikipedia.org/wiki/Experimental_drug
*
10. "Comirnaty" je prva snov na osnovi mRNA v EU, ki jo je Evropska komisija pogojno odobrila kot tako imenovano Covid "cepivo". Tudi drugi dve substanci, ki sta zdaj odobreni kot tako imenovani Covid "cepivi" (proizvajalec: Moderna in AstraZeneca), sta eksperimentalni.
Nezakonito dovoljenje za promet z zdravilom „Comirnaty“ neposredno in osebno vpliva na tožnike/tožnice, saj se grobo krši njihove temeljne pravice do fizične integritete (3. člen Listine EU), visoke ravni varovanja zdravja (člen 168 PDEU, člen 35 EU-listina ) in varstva potrošnikov (člen 169 PDEU, člen 38 Listine EU) z izvedbeno odločbo, kot bo razloženo spodaj.
Posamezni tožniki/ce so že sprejetjem izvedbene odločbe pozvali EU Komisijo in EMO z elektronsko dostavljeno pošto z dne 19.12.2020, da naj ne odobri eksperimentalnih snovi na osnovi mRNA, kot je "Comirnaty", zaradi ogromnih tveganj, ki takrat sploh niso bili v celoti razvidni (glej opozorilno pismo z dne 19. decembra 2020 v dokumentu A.4).
Evropska komisija je 19. januarja 2021 predstavila sporočilo, v katerem je države članice pozvala, naj pospešijo cepljenje po vsej EU z že odobrenimi eksperimentalnimi "cepivi" (najprej s "Comirnaty").
*
11. Zaključek Pfizer študije na živalih: V enem dokumentu so bili povzeti zaključki študije z nasvetom: "Kljub temu ocene imunske okrepivtve, odvisne od cepiva (ADE), ni mogoče ekstrapolati iz živalskih modelov in zahtevajo legitimno raziskavo s poskusi na ljudeh III. faze ali študij na ljudeh." **
https://regenerativemc.com/pfizer-animal-trial-results/
*
12. Republikanska poslanka Marjorie Taylor Greene, tiskovna konferenca 15.6. 2021, ZDA: COVID-19 cepiva nimajo dovoljenja Uprave za hrano in zdravila (FDA), so eksperimentalna. In nekateri ljudje ne želijo biti del eksperimenta cepiv na ljudeh. Zato imajo pravico da odklonijo cepivo in zato ne smejo biti diskreditirani.
*
14. Dr. Biserka Ilin, pedopsihiatrinja: COVID-19 cepiva so eksperimentalna in vsak ki se cepi je udeleženec v 3. fazi kliničnih preizkušanj (od 20.00 min dalje): https://www.youtube.com/watch?v=iCL88IOWvZc
*
15. COMIRNATY® (also known as BNT162b2) receives conditional marketing authorization from the European Commission; this milestone represents a global joint effort to advance the first authorized mRNA vaccine...The vaccine has now been granted a conditional marketing authorization, emergency use authorization, or temporary authorization in more than 40 countries worldwide, including all 27 EU member states: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-receive-authorization-european-union
*
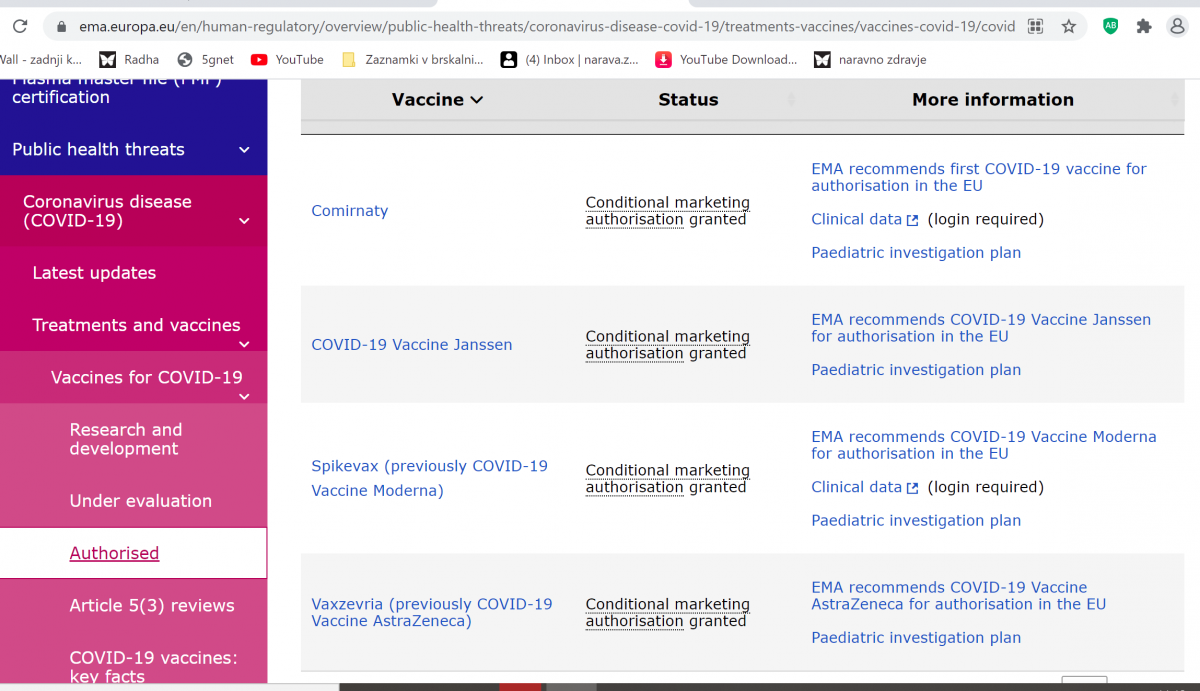
16. DOKAZ EMA: vsa cepiva imajo pogojno dovoljenje za uporabo https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-authorised
*
17. Nadalje, Direktiva Evropskega parlamenta in Sveta Evrope št. 2001/20 v 2 členu točka j) zahteva pisno soglasje pacienta oz. udeleženca v kliničnem preskušanju zdravil in enako v 3. členu 3. točki b in d. Prav tako se pridobitev pisnega soglasja zahteva v navodilih Guidance on the management of clinical trials during the Covid 19 pandemic z dne 4. 2. 2020, verzija 4 v točki 8, stran 10. V Uredbi komisije (ES) št. 507/2006 ki ureja izdajo pogojnih dovoljenj za promet z zdravili v 1. členu 1 točka 10 je dana zahteva da bolnik oz. udeleženec dobi jasne informacije o pogojnosti izdanega dovoljenja1.
Torej, glede na to, da morajo prosilci vseh štirih navedenih cepiv šele v prihodnosti predložiti EMA-i takšne podatke, ki naj bi bili, kot je običajno, potrebni za odobritev njihovega medicinskega preparata, to logično pomeni, da teh podatkov na današnji dan še nimajo (in še manj so jih imeli v času, ko se je pričelo s cepljenjem). Če jih nimajo, to pomeni, da jih zbirajo, če želijo pridobiti dovoljenje za promet, ki ne bo pogojno. In če jih zbirajo to pomeni, da šele poteka raziskava o učinkovitosti njihovega preparata oziroma drugače povedano, gre za eksperimentalno cepivo, ki se daje ljudem.
Vir: Pismo Civilne iniciative slovenskih pravnikov Zdravniški zbornici www.cisp.si
*
Agencija za zdravila JAZMP priznava, da so covid-19 cepiva eksperimentalna, odvetnik Domen Gorenšek zato ugotavlja, da ne morejo biti obvezna:
https://publishwall.si/narava.zdravi/post/613054/zakoni-ne-omogocajo-obveznega-cepljenja-za-covid-19
*
Slovenska zakonodaja eksperimentalno zdravilo imenuje klinično preizkušanje zdravila:
PRAVILNIK
o kliničnih preskušanjih zdravil
I. SPLOŠNE DOLOČBE
1. člen
(področje urejanja in pristojnosti)
(1) Ta pravilnik v skladu z Direktivo 2001/20/ES Evropskega parlamenta in Sveta z dne 4. aprila 2001 o približevanju zakonov in drugih predpisov držav članic v zvezi z izvajanjem dobre klinične prakse pri kliničnem preskušanju zdravil za ljudi (UL L št. 121 z dne 1. 5. 2001, str. 34, z vsemi spremembami; v nadaljnjem besedilu: Direktiva 2001/20/ES) in z Direktivo Komisije 2005/28/ES z dne 8. aprila 2005 o načelih in podrobnih smernicah za dobro klinično prakso v zvezi z zdravili v preskušanju za humano uporabo ter o zahtevah za pridobitev dovoljenja za proizvodnjo ali uvoz takšnih izdelkov (UL L št. 91 z dne 9. 4. 2005, str. 13, z vsemi spremembami; v nadaljnjem besedilu: Direktiva 2005/28/ES) ureja:
- pravice in dolžnosti udeležencev v kliničnem preskušanju zdravila;
- postopek, obliko in vsebino dokumentacije za odobritev ali priglasitev kliničnega preskušanja zdravila;
- postopek, obliko in vsebino dokumentacije za priglasitev pomembnih sprememb kliničnega preskušanja zdravila;
- postopek, obliko in vsebino dokumentacije za obvestilo o zaključku kliničnega preskušanja zdravila;
- postopek obveščanja o resnih neželenih učinkih zdravil v kliničnem preskušanju;
- označevanje zdravil v kliničnem preskušanju;
- shranjevanje dokumentacije o kliničnem preskušanju zdravila;
- nadzor nad kliničnim preskušanjem zdravila.
https://publishwall.si/narava.zdravi/post/584452/kazenska-ovadba-vlade-izraela-zaradi-krsenja-nuremberskega-kodeksa-o-prepovedi-medicinskih-eksperimentov-na-ljudeh-z-uporabo-mrna-cepiva-za-covid-19
*
Evropsko in mednarodno pravo v zvezi s cepivi v kliničnih preskušanjih.
Natančneje, cepiva, ki so na voljo v Franciji, so v tretji fazi kliničnih preskušanj, do 27. oktobra 2022 za Moderno in 2. maja 2023 za Pfizer: do teh datumov so eksperimentalna zdravila [ 3 ], ki se uporabljajo v kliničnem preskušanju [ 4 ] , "vrsta študije: intervencijska (klinično preskušanje)" [ 5 ] , ne glede na število uporabljenih cepiv.
Evropska agencija za zdravila je zato izdala le pogojno dovoljenje za promet (MA) [ 6 ] .
Vendar pa konvencija Oviedo (odstavek 2 člena 26) in Evropska unija zahtevata prosto in informirano privolitev za katero koli zdravilo v fazi kliničnega preskušanja [ 7 ] [ 8 ] .
Obveznost cepljenja se zato zdi "nezakonita" in bi jo sodnik lahko izključil. Opozoriti je treba, da je Evropsko sodišče 24. avgusta 2021 zahtevo gasilcev zavrnilo, vendar je le v nujnih primerih zavrnilo odločitev, ne da bi odločilo o vsebini ( ESČP št. 41950/21 ).
https://publishwall.si/narava.zdravi/post/603986/francoski-odvetniki-zakaj-je-obvezno-cepljenje-kljub-odlocitvi-ustavnega-sveta-nezakonito
*
*
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7096781/
*
Reuters svojega zanikanja, da so cepiva eksperimentalna, ni utemeljil z zakoni in pravnimi dejstvi: https://www.reuters.com/article/factcheck-covid-vaccines-idUSL1N2M70MW
*
-
Pismo je posredoval dr. Andraž Teršek: "Koliko posameznikov res pozna svoje temeljne pravice in ugotovim, da je največje zlo človeštva neznanje, ignoranca, slepo zaupanje v ljudi pomanjkanje ljubezni. Misel, da vsi ljudje delajo za skupno dobro je naivno dejanje. Misliti ne pomeni nič vedeti, ker najprej je bila misel, potem šele fizična manifestacija te misli(biblija). Cepiva so v testni fazi do leta 2023. Agenda katera se izvaja, se bo izvajala do leta 2030. V cepivu so snovi, katere so nevarne človeškemu bitju, že, če so v hrani, ne pa, da si jih vbrizgamo v telo. Snov, ki je v teh cepilih in je nevarna je Rèkombinántna DNK je sintezna molekula DNK, sestavljena iz fragmentov DNK različnega izvora – gre za kombinacijo DNK-zaporedij, ki se v takšni obliki naravno ne nahajajo.( Gre za konbinacijo dveh različnih bitij). Pri tehnikah genetske modifikacije rekombinantno DNK proizvedejo z vstavljanjem določenega DNK-zaporedja v obstoječi genom nekega organizma v drug organizem ( s cepljenjem). To izvedejo na primer s pomočjo bakterijskega plazmida, ki ima vlogo vektorja, torej prenašalca zaporedja, ki ga želimo vstaviti v telo.
____
4. Cepiva so po definiciji eksperimentalna. V fazi 3 testiranj bodo ostala do leta 2023. Prejemniki so osebe, ki so upravičene do informiranega soglasja po Nürnberškem kodeksu in drugih zaščitah, vključno s Resolucijo Parlamentarne skupščine Sveta Evrope 2361 [43] in pogoji Uprave za hrano in zdravila (FDA) o odobritvi za uporabo v sili [29]. V zvezi z varnostnimi podatki iz faze 1 in 2 preskušanj, kljub sprva velikim vzročnim skupinam, revija Vaccine poroča, da je "strategija cepljenja, izbrana za nadaljnji razvoj, lahko bila dana zgolj 12 udeležencem testiranja."[32].
S tako zelo majhnimi velikostmi vzorcev, ugotavlja revija, bodo potrebne "večje študije faze 3, izvedene v daljšem časovnem obdobju", da bi se vzpostavila varnost. Tveganja, ki jih je treba oceniti v preskušanjih faze 3 do leta 2023, pri celotni populaciji kot preiskovancih, ne vključujejo le tromboze in krvavitev, ampak tudi druge avtoimunske odzive, alergijske reakcije, neznane tropizme (destinacije v tkiva) lipidnih nanodelcev [35], okrepitev bolezni v odvisnosti od protiteles (ADE) [43-46] in vpliv naglih, vprašljivo izvedenih, slabo reguliranih[47] in neskladnih proizvodnih metod s prenosom tveganja zaradi potencialno škodljivih nečistoč, kot so nenadzorovani ostanki DNK [48]. Cepiva niso varna niti za prejemnike niti za tiste, ki jih dajejo ali dovolijo njihovo uporabo.
*
Zaslišanje pediatrinje dr. Angeline Farella v Senatu Texasa (7.40): Proizvajalci niso izvedli testiranja na živalih, ker so umirale. Zdaj so ljudje poskusni zajčki eksperimentalnih cepiv:
*
https://www.delo.si/avtor/alojz-ihan/
dr. Ihan RTV intervju: https://www.google.com/search?q=Dr+Ihan+RTV+intervju&rlz=1C1GCEA_enSI871SI871&oq=Dr+Ihan+RTV+intervju&aqs=chrome..69i57j69i64.5647j0j15&sourceid=chrome&ie=UTF-8

Miha Možina FB 24.11. 2021: G. Alojz Ihan. Človek študira, gara dolga leta da postane zdravnik, specialist klinične mikrobiologije, predstojnik Katedre za mikrobiologijo in imunologijo in redni profesor mikrobiologije in imunologije na Medicinske fakulteti Univerze v Ljubljani. Nato v 59-tem letu življenja, na višku svojih ustvarjalnih moči, ko je dosegel že praktično vse, ko ima za sabo že več kot 120 objav znanstvenih člankov v najrazličnejših mednarodnih znanstvenih revijah, napiše kolumno, ki jo je objavil časnik DELO, 21. avgusta 2020 (glej spodaj). Potem ga angažira vlada in naenkrat nič od tega, v kar je verjel še leto poprej, ne velja več. Zaupajmo stroki.
*
https://www.ema.europa.eu/en/documents/other/pharmacovigilance-plan-eu-regulatory-network-covid-19-vaccines_en.pdf
*
https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation
*
https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-development-evaluation-approval-monitoring#accelerated-evaluation
*
https://vaccination-info.eu/en/covid-19/covid-19-vaccines
*

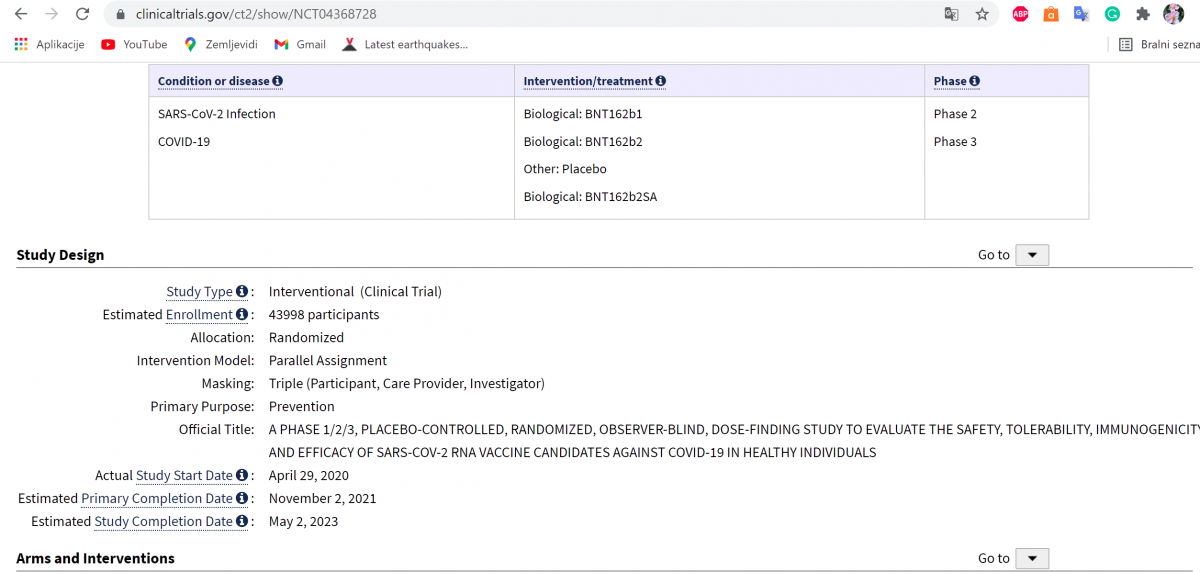
This is a Phase 1/2/3, randomized, placebo-controlled, observer-blind, dose-finding, vaccine candidate-selection, and efficacy study in healthy individuals.
The study consists of 2 parts: Phase 1: to identify preferred vaccine candidate(s) and dose level(s); Phase 2/3: an expanded cohort and efficacy part.
The study will evaluate the safety, tolerability, and immunogenicity of 3 different SARS-CoV-2 RNA vaccine candidates against COVID-19 and the efficacy of 1 candidate:
- As a 2-dose (separated by 21 days) schedule;
- At various different dose levels in Phase 1;
- As a booster;
- In 3 age groups (Phase 1: 18 to 55 years of age, 65 to 85 years of age; Phase 2/3: ≥12 years of age [stratified as 12-15, 16-55 or >55 years of age]).
The candidate selected for efficacy evaluation in Phase 2/3 is BNT162b2 at a dose of 30 µg.
Participants who originally received placebo will be offered the opportunity to receive BNT162b2 at defined points as part of the study.
In order to describe the boostability of BNT162, and potential heterologous protection against emerging SARS-CoV-2 VOCs, an additional dose of BNT162b2 at 30 µg will be given to Phase 1 participants approximately 6 to 12 months after their second dose of BNT162b1 or BNT162b2. This will provide an early assessment of the safety of a third dose of BNT162, as well as its immunogenicity.
The assessment of boostability will be further expanded in a subset of Phase 3 participants at selected sites in the US who will receive a third dose of BNT162b2 at 30 µg or a third and potentially a fourth dose of prototype BNT162b2VOC at 30 µg (BNT162b2s01, based upon the South African variant and hereafter referred to as BNT162b2SA). A further subset of Phase 3 participants will receive a third, lower, dose of BNT162b2 at 5 or 10 µg.
To further describe potential homologous and heterologous protection against emerging SARS-CoV-2 VOCs, a new cohort of participants will be enrolled who are COVID-19 vaccine-naïve (ie, BNT162b2-naïve) and have not experienced COVID-19. They will receive BNT162b2SA given as a 2-dose series, separated by 21 days.
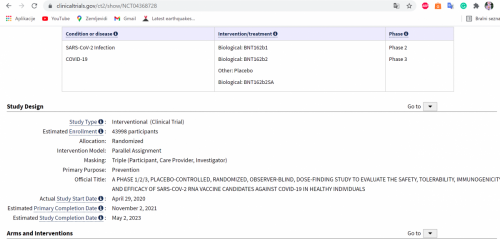
https://clinicaltrials.gov/ct2/show/NCT04368728
*
Zaslišanje v avstralijskem Senatu: Zakaj so Covid-19 cepiva eksperimentalna? Ker niso znani dolgoročni in transgeneracijski učinki, niti učinkovistos cepiva pri preprečevanju prenosa okužbe:
https://www.facebook.com/marija.stoj.1/videos/879646146301523
*
V primeru pogojnega dovoljenja za promet (CMA) EMA temeljito oceni vse potrebne informacije, da potrdi, da koristi odtehtajo tveganja zdravila.
Poleg podatkov, ki dokazujejo varnost in učinkovitost, morajo na primer podatki, predloženi v vlogi za pridobitev dovoljenja za promet s cepivom COVID-19, vključevati informacije o:
skupini ljudi, ki bodo prejeli cepivo;
farmacevtski kakovosti in čistosti cepiva;
proizvodnji in nadzoru serij;
skladnosti z mednarodnimi zahtevami za laboratorijsko testiranje in izvajanje kliničnih preskušanj;
vrstah imunskih odzivov;
neželeni učinki, npr. pri starejših ljudeh ali nosečnicah;
označevanje in navodilo za uporabo;
način obvladovanja in spremljanja tveganj, ko bo cepivo odobreno.
Postopek pogojnega dovoljenja za promet daje državljanom potrebno zagotovilo, da se pri izvajanju programov množičnega cepljenja izvajajo vse zahteve, vključno z neodvisnim nadzorom serij cepiva, ki ga izvaja mreža uradnih laboratorijev za nadzor zdravil (OMCL), preden cepivo pride do bolnika.
Če država članica izda dovoljenje za uporabo v nujnih primerih, se sama odloči, katere podatke potrebuje za takšno dovoljenje in katere zahteve bo uvedla za uporabo in nadzor cepiva. Zato je mogoče, da se zahtevajo manj podrobni podatki in da se naložijo manj stroge obveznosti kot v postopku pogojnega dovoljenja za promet, ki se nanašajo na primer na:
-postopkom izdelave cepiva;
-certificiranjem statusa dobre proizvodne prakse (GMP) objektov, v katerih se bo cepivo proizvajalo;
-obsegom kliničnih podatkov, vključno z velikostjo zbirke podatkov o varnosti in analizami kliničnih podatkov o učinkovitosti, ki so na voljo.
Medtem ko popolna uporaba določb, ki jih zakonodaja EU posebej predvideva za nujne primere, pospešuje postopek odobritve, lahko trdnost ocenjevalnega okvira CMA in obseg podatkov, ki jih oceni EMA, povzročita, da postopek zahteva več časa kot postopek odobritve uporabe v nujnih primerih.
*
Po pozitivni oceni Evropske agencije za zdravila (EMA) o njihovi varnosti, kakovosti in učinkovitosti je Komisija doslej izdala pogojno dovoljenje za promet s štirimi cepivi, ki so jih razvili:
BioNTech in Pfizer, 21. decembra 2020
Moderna, 6. januarja 2021
AstraZeneca, 29. januarja 2021
Janssen Pharmaceutica NV, 11. marca 2021
*
V pogodbi proizvajalca Pfizer je navedeno (3.3 c):
Kupac se slaže da cjepivo neće krenuti u serijsku proizvodnju. Cjepivo ostaje eksperimentalno, Kupac priznaje da je svjestan kako dugoročni učinci i djelotvornost cjepiva nisu poznati, te da mogu postojati štetni učinci.
Pfizer nije odgovoran ni za što, vezano uz njegovo eksperimentalno Covid cjepivo, prodaje ga uz sigurnu naplatu pod uvjetima koje on diktira. U Ugovoru je navedeno kako kupac „mora zaštititi Pfizer od odgovornosti za potraživanja i gubitke, to mora provesti putem zakonskih ili regulatornih zahtjeva, a o dovoljnosti takvih napora odlučuje Pfizer“.
Nuremberg Code Against Nazi Experiments Should Stop Further COVID-19 Vaccinations
By Peter R. Breggin MD and Ginger R. Breggin
This report examines COVID-19 vaccinations in light of the principles established by the Nuremberg Code, an ethical standard for scientific research developed by the judges in the Nazi War Crimes Trials after World War II. The Nuremberg Code (1949) (wayne.edu)
The Kaimowitz Trial
In 1972, I was the lead medical and psychiatric expert in a three-judge trial called Kaimowitz v. Department of Mental Health that stopped lobotomy and psychosurgery (psychiatric brain mutilation) from being imposed on state mental hospital patients in Michigan, even if the inmates said that they consented. The injunction was brought by attorney Gabe Kaimowitz on behalf of John Doe and the Medical Committee for Human Rights. The judges’ decision led to the termination of all psychiatric surgery in state and federal facilities in the United States. It became the first of many victories in my campaign to stop the worldwide resurgence of psychiatric brain mutilation.
The three judges determined that inmates of a highly authoritarian, oppressive institution are in no position to freely give consent to dangerous experiments because of their demoralizing circumstances and the inherent threat of coercion, retaliation, pressure, and threats.
These legal and moral principles are embedded in the Nuremberg Code and derived from the experiences of inmates of Nazi concentration and extermination camps who supposedly volunteered for dangerous experiments. My testimony prepared the way for the judges by describing the history and current conditions of state mental hospitals enabling them to compare them to concentration and extermination camps. It also demonstrated that the surgery was indeed mutilating to the brain and destructive to all higher mental and emotional functions.
Applying the Nuremberg Code to COVID-19 Vaccinations
Recently, Ginger Breggin emailed documents and my publications about the Kaimowitz case to a group of leading physicians and lawyers who are defending human rights from COVID-19 fraud and oppression. I thought it was a good idea because the Kaimowitz decision and the Nuremberg code apply to giving COVID-19 vaccinations to supposedly consenting inmates of jails, prisons, nursing homes and other coercive institutions where they are at the mercy of their keepers and healthcare providers.
But Ginger had a broader perspective. We are all living in an oppressive society in which if we refuse vaccinations we will be subjected to social and governmental pressure, as well as extreme fear tactics. Even worse, many institutions, such as schools and public events, are already announcing that they will not admit individuals who lack vaccine passes. People in the government are warning about vaccine passes being used to control domestic and international travel. Vaccine passports are weapons of social control.
Public health policies and practices under Anthony Fauci were used to engorge the wealth of billionaires and huge corporations at the expense of humanity. As an even greater long-term menace, COVID-19 public health measures are being used to ramp up and enforce top-down government in America and throughout the world. These, and all the other observations made in this report, are documented in detail in our book, COVID-19 and the Global Predators.
Even if not all the new threats materialize, they are already sufficient to intimidate millions of people into taking vaccinations. Many people, for example, are leery of the vaccinations but accept them out of fear of the legal, political, and social repercussions of refusing. No one living in America today, whether in an institution or at home, is sufficiently free of coercion to voluntarily accept COVID-19 vaccines.
The vaccines being pushed on Americans are vastly increasing the wealth and power of the pharmaceutical industry and their investors like Bill Gates. None have gone through the regular FDA approval process. All have been rushed through. All the vaccines are dangerous and extremely experimental. Reports of vaccine-related deaths to the CDC reporting systems called VAERS are mounting and will probably exceed 2,000 in the United States when this report is published. Yet these voluntary reports represent only a small fraction of the true numbers. Physicians are always hesitant to report deaths that might be related to their treatments and many physicians are not even aware that their patients have been vaccinated. The healthcare providers who administer the vaccinations typically have no follow up information about the next 48 hours during which many vaccinated patients will die. Reports of deaths in such great numbers in association with a medicine or vaccine would usually result in a stoppage but the CDC is refusing to do a thorough evaluation of the reports. Many other countries are already halting the administration of at least one of the COVID-19 vaccines.
Stop the COVID-19 Vaccinations Now!
As my recent scientific publications on vaccines confirms, I am not against all vaccinations or vaccinations in principle. But the COVID-19 vaccinations must be stopped. The vaccines are unprecedented in their mechanism, dangerous and very experimental, and the reported deaths are mounting. There is insufficient information for anyone to go give informed consent. And no one in America today is free enough from misinformation, coercion, and threats to make a freely informed decision. Under the principles of the Nuremberg Code, as well as general medical ethics which it reflects, the government should be prohibited from experimenting on its citizens with COVID-19 vaccines.
The tens of millions of people already vaccinated need to be evaluated for both safety and effectiveness, a mammoth task that will take considerable time and effort. When accurate information about the vaccines can be communicated to the public and when citizens are no longer living under such coercive conditions, it may become possible for individuals to make informed freely-chosen decisions about taking the vaccine. But that is a long way off. Before the vaccinations can resume, we Americans must retake our individual and political freedom.
Peter R. Breggin MD is known as “the Conscience of Psychiatry” for his decades of research, forensic work, reform efforts, and many scientific and popular publications. Ginger R. Breggin, his wife and partner in all these activities, has coauthored several books with him, including their newest book, COVID-19 and the Global Predators: We Are the Prey.
https://breggin.com/nuremberg-code-against-nazi-experiments-should-stop-further-covid-19-vaccinations
*
Zavajanje agencija JAZMP: »To pomeni, da so cepiva proti covidu-19, ki so trenutno v uporabi, pridobila dovoljenje za redno uporabo, ki velja za eno leto in se lahko obnovi, v kolikor bo imetnik dovoljenja za promet izpolnil obveznosti v predpisanih rokih.«
II — The planned studies are still in progress and are spread over a period ranging from “2021 to at least 2024”
All of the studies submitted during the MA application are summarized in the EPAR (European Public Assessment Report). This report is published on the European Medicines Agency (EMA) website. The planned studies, not yet completed, are also included.
This schedule, which “extends from 2021 to at least 2024,” depending on which COVID-19 vaccine is involved, is defined in the “annexes” of the conditional marketing authorization and in the published EPARs.
As an example, the BioNTech/Pfizer vaccine received this European conditional MA on December 21, 2020. And the deadline for filing “confirmation” of efficacy, safety, and tolerability of this vaccine is “December 2023.”
The Moderna vaccine was granted marketing authorization on January 6, 2021. The deadline for filing “confirmation” of efficacy, safety, and tolerability of the vaccine is “December 2022” at the earliest.
AstraZeneca’s vaccine was granted marketing authorization on January 29, 2021. The deadline for filing “confirmation” of efficacy, safety, and tolerability of the vaccine is “March 2024.”
The Janssen vaccine was granted conditional European marketing authorization on March 11, 2021. The deadline for submitting “confirmation” of the vaccine’s efficacy, safety and tolerance is “December 2023.”
However, to date — and this is undoubtedly where the unprecedented and exclusive revelation of this study lies — another deadline has been set for these four vaccines. This deadline no longer concerns only the ongoing clinical trials, but also the “proof of quality for the active substance and the finished product” itself: that is, the intrinsic quality (the heart) of the product sold and administered to millions of people.
III — The published official documents also underline the incompleteness of the evidence concerning the “quality” of the “active substance” and “excipients,” the “manufacturing process,” the ”reproducibility of the batches marketed”, etc.
The deadline for submitting additional evidence on the “quality” of the “active substance” and the “finished product” (i.e., the vaccine that is authorized and sold) is set for:
- “July 2021” for BioNTech/Pfizer;
- “June 2021” for Moderna;
- “June 2022” for Astra Zeneca;
- “August 2021” for Janssen.
Indeed, for these 4 vaccines, paragraph E, “Specific obligation regarding post-authorization measures for the conditional marketing authorization,” taken from Annex II of the MA, clearly states the following:
For the BioNTech/Pfizer vaccine (pages 18-19)
By “March 2021,” the laboratory must provide “additional validation data” to “confirm the reproducibility of the finished product manufacturing process.”
By “July 2021,” the laboratory must provide missing information to:
-
- “complete the characterization of the active substance and the finished product;”
- “strengthen the control strategy, including the specifications of the active substance and the finished product” in order to “ensure the constant quality of the product;”
- “provide additional information regarding its synthesis process and control strategy” in order to “confirm the purity profile of the excipient ALC-0315” and “to ensure quality control and batch-to-batch reproducibility throughout the life cycle of the finished product;”
- and by “December 2023,” and “in order to confirm the efficacy and safety” of this vaccine, the company “shall submit the final clinical study report for the randomized, placebo-controlled, blind observer study (Study C4591001).
For the Moderna vaccine (page 15)
The laboratory should provide the missing information to:
-
- “complete the characterization of the manufacturing processes of the active substance and the finished product” (deadline “January 2021”);
- confirm the reproducibility of the manufacturing process of the active substance and the finished product (initial and final batch sizes) (deadline “April 2021”);
- “provide additional information on the stability of the active substance and the finished product and review the specifications of the active substance and the finished product after longer industrial practice” with the aim of “ensuring consistent product quality” (deadline “June 2021”);
- “submit the final study report for the randomized, placebo-controlled, blinded clinical trial for the mRNA-1273-P301 observer” to “confirm the efficacy and safety of COVID-19 vaccine Moderna” (by December 2022).
For the Astra Zeneca vaccine (pages 14-15)
The laboratory must submit the missing information in order to:
-
- “provide additional validation and comparability data, and initiate further testing” with the aim of “confirming the reproducibility of the manufacturing processes of the active substance and the finished product” (by “December 2021”);
- “Provide the main analysis (based on the December 7 data cut-off (post database lock) and the final analysis of the combined pivotal studies” to “confirm the efficacy and safety of COVID-19 Vaccine AstraZeneca” (deadline “March 5, 2021” (for the main analysis) and “May 31, 2022” (for the combined analysis);
- “submit final reports of the randomized controlled clinical studies COV001, COV002, COV003 and COV005” to “confirm the efficacy and safety of COVID-19 Vaccine AstraZeneca” (due “May 31, 2022”);
- “provide additional data regarding the stability of the active substance and the finished product and revise the specifications of the finished product after extensive industrial practice” in order to “ensure consistent product quality” (deadline “June 2022”);
- “submit the synthesis and summaries of the primary analysis and the final clinical study report for study D8110C00001” to “confirm the efficacy and safety of COVID-19 vaccine AstraZeneca in the elderly and in subjects with underlying disease” — due “April 30, 2021” (for the primary analysis) and “March 31, 2024” (for the final study report).
For the Janssen vaccine (page 18)
The laboratory should submit the missing information to:
-
- “provide additional comparability and validation data” to “confirm the reproducibility of the manufacturing process of the finished product” (deadline “August 15, 2021”);
- submit the final report of the VAC31518COV3001 randomized, placebo-controlled, single-blind clinical study to “confirm the efficacy and safety of the COVID-19 Ad26.COV2.S vaccine” by December 31, 2023.
These facts allow us to offer a conclusion.
EMA Conclusion
For these reasons, which are not exhaustive, it has proved useful to look for and read the content of the paragraph E: “Specific obligation relating to post-authorization measures concerning the conditional marketing authorization,” extracted from Annex II of the MA, corresponding to each of these 4 vaccines against COVID-19.
The inadequacy of the evaluation does not only concern the clinical trials (studies conducted in humans (women and men)), but also the quality of the active substance, the excipients (some of which are new) the manufacturing process, and the batches released and administered to humans in several countries around the world.
Moreover, these new excipients must be considered as new active ingredients, and thus be the subject of a complete evaluation file similar to that required for a new active ingredient.
Changing the commercial name of one of these vaccines, as was recently announced for the AstraZeneca vaccine in particular, can only be considered as a cosmetic arrangement of the product’s image for marketing purposes (winning new public confidence, boosting sales). It would not answer the questions raised concerning the quality, efficacy and safety of the product. This is one of the usual techniques used to put make-up on (dissimulate) certain undesirable characteristics of the product concerned. It is a technique that has been used to present other drugs in the best possible light.
As already mentioned, in the field of medicines (including vaccines), the “release” of the finished product (intended for sale) is the final stage of control (of quality and therefore of safety) before making these products available to the population.
This key stage of “release” of batches is the pharmaceutical responsibility of the manufacturers. However, the responsibility of the users (institutions and health professionals in particular) may also be involved.
In our opinion, these clinical studies should never have begun before the intrinsic quality of the finished product and its manufacturing process had been fully mastered; before the formulas of these vaccines had been stabilized.
How can the results of these clinical trials, conducted on a global scale, be compared if the vaccine administered can vary from one manufacture to another, from one batch to another, from one region to another?
These variabilities, which impact the very core of the product, could even invalidate any clinical trials conducted.
Even in the case of a health emergency, it is therefore difficult for us to understand the basis for the MA (marketing authorization) that has been granted to these COVID-19 vaccines.
In addition to the uncertainties related to COVID-19, there are also the approximations related to the use, and the intrinsic quality, of these vaccines. Now two problems will have to be managed instead of one.
The maneuver seems subtle. The useful information is available in the official documents published in the framework of the MA; but this data is not made visible by the official discourse. It seems the latter has only tried to present these products as being effective and safe, without reservations; even though the formulas and manufacturing processes of these vaccines do not even seem to have been fully stabilized yet.
These new revelations, which are undoubtedly unprecedented and exclusive, further cast doubt on the validity of consent (a fundamental freedom) that is supposed to be free and informed, and which is said to have been given by the people who are now already vaccinated.
Every person has the right to clear, fair and appropriate information. This information is also perennial: if new data is revealed, those already vaccinated must be informed a posteriori (after the administration of this or that vaccine).
The “obligation” to vaccinate cannot therefore be sustained, even in a disguised form, notably through a “vaccine passport.”
This new analysis further confirms our previous reflections such as the one entitled “Could the Covid-19 vaccine (Tozinameran; COMIRNATY°) be qualified as ‘defective’ by a judge?”. Read here.
Or those expressed in the two open letters that have already been sent to the Minister of Solidarity and Health and to the seven Orders of health professionals.
https://int.artloft.co/why-did-a-french-drug-assessment-center-demand-removal-of-all-four-widely-used-covid-vaccines/
*
Ali bi lahko sodnik cepivo proti Covid-19 (Tozinaméran; COMIRNATY °) označil za "okvarjeno"?
» Učinkovitost cepiva bo najprej odvisna od naše sposobnosti krepitve zaupanja . Odgovoriti na vprašanja, odpraviti dvome, zmanjšati strahove, na to se moramo osredotočiti v prihodnjih tednih. Na vseh ozemljih v vsaki občini so zdravniki in zdravstveni delavci mejnik, spoštovana osebnost, katere glas poslušajo in slišijo. Tesni odnosi, ki vas vežejo na naše sodržavljane, so edinstveni. " (Minister za solidarnost in zdravje; v " PORTFOLIO ", priloženem " Vodniku po cepljenju za zdravnike, medicinske sestre in farmacevte ", objavljenem 31. decembra 2020)
Ne, učinkovitost cepiva je najprej odvisna od podatkov, potrjenih v kliničnih preskušanjih (izvedenih pri ljudeh). Vloga zdravstvenega delavca je predvsem zagotavljanje jasnih, poštenih in ustreznih informacij za pridobitev prostega in informiranega soglasja osebe; kot mu nalagajo zlasti kodeks javnega zdravja in pravila njegovega reguliranega poklica.
Zdi se, da nas upravljanje Covid-19, povezano s Sars-CoV-2, popelje v obdobje post-znanosti in post-prava. Primer cepiva proti Covid-19 bi lahko ponazoril to ugotovitev.
Ta razmislek vam ponuja razmišljanje v treh stopnjah, kot bi to storil sodnik: prvič, slednji opredeljuje pravno državo (I) ; nato to pravilo sooči z dejstvi subjekta (vrste: tukaj cepivo proti Covid-19) (II) ; in nazadnje naredi zaključek (reši spor) (III) .
I- Kaj je po zakonu "okvarjen" izdelek?
Cepivo je zdravilo. Je "izdelek" v smislu civilnega zakonika.
Na splošno se v primeru škode zaradi neželenih učinkov, za katere se domneva, da so medicinskega izvora, sodnik zlasti opira na nekatere določbe tega civilnega zakonika, da prevzame odgovornost proizvajalca (farmacevtskega laboratorija) tega "okvarjenega" izdelka .
Žrtev mora dokazati številne elemente, vključno z "napako" izdelka. Po civilnem zakoniku je izdelek okvarjen, če "ne ponuja varnosti, ki bi jo lahko upravičeno pričakovali" . Nevarnost izdelka je lahko na različnih ravneh: oblikovanje, izdelava, uporaba itd. Sodnik upošteva nekatera merila, kot so predstavitev izdelka, uporaba, za katero je namenjen itd.
Več sodnih odločb (sodna praksa) posebno pozornost namenja navodilom, ki so priložena izdelku. Nezadostne informacije o tveganjih zdravila (cepiva) omogočajo sodniku, da ugotovi omenjeno napako tega zdravila. Tudi če je tveganje omenjeno v tem navodilu, se lahko odgovornost proizvajalca obdrži zlasti, če zdravilo predstavlja neugodno razmerje med koristjo in tveganjem (tveganje je večje od koristi).
Na posebnem področju cepiv in od leta 2008 se zdi, da je najvišja sodna pristojnost (kasacijsko sodišče) svojo sodno prakso premaknila v smer, ki je ugodnejša za žrtev: napako je mogoče dokazati s preprosto domnevo, če temelji o "resnih, natančnih in skladnih indeksih"na primer časovna bližina med injiciranjem cepiva in pojavom neželenih učinkov; odsotnost osebne in družinske anamneze v zvezi s temi stranskimi učinki; obstoj znatnega števila opisanih in objavljenih primerov, ki poročajo o teh škodljivih učinkih po dajanju omenjenega cepiva ... Sodišče Evropske unije (Sodišče Evropskih skupnosti) je leta 2017 to možno uporabo dokazov potrdilo z domnevo. Laboratorij lahko predloži nasprotni dokaz. Kljub temu dogajanju je položaj sodnikov še negotov. Žrtev je lahko razočarana.
II- Uporaba te pravne države za cepiva proti Covid-19
V tem primeru se je treba zlasti na primeru cepiva iz laboratorijev Pfizer / BioNTech (Tozinaméran; COMIRNATY °) vprašati, ali bi sodnik lahko ocenil , da je ta izdelek "okvarjen" . Isto vprašanje in enaka utemeljitev veljata tudi za druga cepiva proti Covid-19. Ta analiza je omejena na primer cepiva (Tozinaméran; COMIRNATY °), ki je prvi koristil „pogojno“ dovoljenje za promet (MA), zlasti v Franciji; in za katere imamo zato daljše spremljanje (v primerjavi z drugimi cepivi).
II.1- Regulator promovira urnik cepljenja, ki se razlikuje od urnika cepljenja, ki ga odobri dovoljenje za promet
7. januarja 2021, je varnostna agencija nacionalni drog (ANSM), sam, ki se zavzema za off-label uporabi tega cepiva. Dejansko je v svojem " mnenja (...) v zvezi z drugim odmerkom cepiva Comirnaty od Pfizer-BioNtech - informacijske točke" je ANSM meni, da je "rok za upravljanje 2 nd mogoče predvideti odmerek med 21 in 42 dni «Ob tem, da spomnimo, da je to cepivo » odobreno v Evropi « za » urnik dajanja «, ki » temelji na dveh odmerkih, ki sta v razmiku najmanj 21 dni «. Regulatorni organ, žandar za zdravila, si zato dovoljuje, da spremeni urnik cepljenja (odmerek zdravila), kot ga je odobrilo dovoljenje za promet. Zdi se, da tak odstop od MA potrjuje proizvajalec sam. Farmacevtsko podjetje se zato zdi previdnejše od regulatorja: laboratorijska datoteka Pfizer / BioNTech kaže na okrepitev 21 dni po prvi injekciji. ANSM je sledil nasvetom Evropske agencije za zdravila. Slednji opozarja, da je za takšen premik odgovoren predpisovalec. WHO (Svetovna zdravstvena organizacija) odobri ta premor.
Vendar pa je še več. Na področju, je v pojasnilu z dne 8. januar 2021 in razpošilja vse osebje, bolnišnica center ponuja svoj urnik cepljenja: uporaba dveh odmerkov razporejene na vsaj 19 dni narazen " . 5. januarja 2021 je po objavljenem cepljenju profesorja medicine, vodje oddelka v univerzitetnem bolnišničnem centru (CHU), tisk zapisal: "Vodja oddelka bo v desetih dneh prejel še eno injekcijo, tako da bo imunizacija skupaj " .
Drugi regulatorji, druge države, spoštujejo MA
Nemčija noče iti izven okvira MA in se je odločila, da ne bo odložila dajanja druge injekcije.
Ameriški regulativni organ (FDA: uprava za hrano in zdravila) meni, da ta uporaba, ki ni označena, ne temelji na nobenih razpoložljivih in zanesljivih podatkih.
Neznane posledice: možna tveganja
Taka sprememba urnika cepljenj otežuje odgovor na postavljena vprašanja: kakšna zaščita med obema injekcijama? Kako dober je imunski odziv po drugem odmerku?
Možna tveganja so lahko naslednja: bolezen, ki se poslabša s cepljenjem, avtoimunski stranski učinki, razvoj odpornosti virusa (Sars-CoV-2) na cepivo itd.
II.2- Vzorec s 6- th odmerek pa se vsebino čaše potrjene 5 odmerkov na NMA
Zdi se, da je bila promovirana še ena svoboda. Nekateri priporočajo cepljenje 6 oseb z vialo, ki je po podatkih dovoljenja za promet z zdravilom namenjena cepljenju 5 oseb. Vendar pa je preostala količina (kot bi nekateri uporabljajo za omenjene 6 th odmerek) omogoča zbiranje, natančno, je 5 rednih odmerkih in še posebej zadnje (5 th ) odmerek. Ta preostali volumen omogoča izpiranje igle in odstranitev zračnih mehurčkov iz brizge.
II.3- Vprašljivi, nepopolni ali celo netočni podatki, razširjeni prek dokumentov, ki so jih pripravili začasni organi
Napake
Na primer glede načina dajanja cepiva po intramuskularni (IM) poti je "PORTFOLIO", priložen " Vodniku po cepljenju za zdravnike, medicinske sestre in farmacevte" , objavljen 31. decembra 2020 na spletni strani ministrstva solidarnosti in zdravja, označuje: " Naredite kožno gubo med palcem in kazalcem" .
Toda v svoji različici z dne 5. januarja 2021 ta "PORTFOLIO" vsebuje popravek tega navodila: "Močno raztegnite kožo med kazalcem in palcem, ne da bi naredili kožno gubo " .
Na dan 31. decembra 2020 je ta dokument vseboval 61 strani. 5. januarja 2021 vsebuje le 26 strani.
Vprašljivi podatki o učinkovitosti cepiva
Na primer, v "listu 2 INFORMACIJE ZA REZIDENTE V OBSTAVIH ZA STAREJŠE IN NJIHOVE DRUŽINE" navedenega "PORTFOLIO" je navedeno naslednje:
"1. Zakaj se cepiti proti COVID-19? : Cepljenje proti COVID-19 vas bo zaščitilo pred zapleti in pojavom hudih oblik te bolezni. Študije so pokazale, da je cepivo zelo učinkovito pri zaščiti pred okužbami. "
V resnici pa je CTIAP, tako kot neodvisni pregled Prescrire , opazil "negotovost" glede učinkovitosti tega cepiva na omenjene "resne" oblike Covid-19 (glej članek z dne 26. decembra 2020).
Kot je prikazano v poročilu z dne 7. decembra 2020 o sestanku, ki ga je vodil delegat minister, pristojen za avtonomijo, je takšna učinkovitost predstavljena v obliki hipoteze, ki jo je treba dokazati: "to cepivo za permett bi močno zmanjšalo nevarnost resne oblike zaradi okužbe s COVID19 " . Besede imajo pomen; konjugacija glagola ima pomen: uporaba pogojnika je oblika previdnosti. To poročilo je bilo omenjeno med konferenco CTIAP 17. decembra 2020.
Ameriški regulativni organ (FDA: hrana in zdravila) je ugotovil, da rezultati kliničnih študij ne izpolnjujejo meril uspešnosti, ki so bili predhodno opredeljeni za hude oblike.
Avtorji v nasprotju trditvi so pozvane, da predložijo dokazilo o učinkovitosti cepiva, ki dokazuje, da znižanje v bolnišnico , od posledic do bolj ali manj dolgoročno, del življenjsko nevarne (bivanje v intenzivni) od smrti ... In prenos virusa ...
Nepopolni podatki o neželenih učinkih
Na primer, v tem istem "listu 2 INFORMACIJE ZA REZIDENTE V OBSTAVIH ZA STAREJŠE IN NJIHOVE DRUŽINE" je navedeno naslednje:
"4. Ali obstajajo kakšni stranski učinki tega cepljenja? Kot pri vseh cepivih se lahko po cepljenju pojavijo neželeni učinki: bolečina na mestu injiciranja, utrujenost, glavobol, bolečine v mišicah ali sklepih, nekaj mrzlice in rahlo zvišana telesna temperatura. Te motnje hitro izginejo. "
V resnici je ta seznam nepopoln, kar dokazujejo zlasti naslednji dokumenti:
Na spletni strani ANSM sta na voljo dva dokumenta
Povzetek glavnih značilnosti zdravila ( SPC ) in navodilo za uporabo, izdelano v okviru dovoljenja za promet z zdravilom (prim. Datoteke PDF, ki so na voljo v prikazani tabeli );
List "Neželeni učinki cepiva Pfizer / BioNTech COMIRNATY °: kaj morate vedeti" (na dnu strani, za isto tabelo).
Sporočilo za javnost Haute Autorité de Santé (HAS) z dne 24. decembra 2020 : kliknite tukaj
Članek v reviji Prescrire z dne 23. decembra 2020 : kliknite tukaj
Konferenca CTIAP 17. decembra 2020 : kliknite tukaj
Kodeks javnega zdravja in sodna praksa zahtevata informacije o pogostih ali resnih, celo izjemnih, neželenih učinkih .
II.4- Farmakovigilanca zaradi premajhnega obveščanja, ovir in nekaznovanosti
Na primer, in še vedno v istem "listu 2 INFORMACIJE ZA REZIDENTE V OBSTAVIH ZA STAREJŠE IN NJIHOVE DRUŽINE" je navedeno naslednje:
"V okviru nacionalne kampanje cepljenja proti COVID-19 Nacionalna agencija za varnost zdravil (ANSM) vzpostavlja poseben sistem za okrepljeno spremljanje škodljivih učinkov cepiv proti Covid-19 na francoskem ozemlju. "
8. januarja 2021 je ANSM objavil " Posodobitev stanja o nadzoru cepiv proti COVID-19", v kateri je zlasti navedeno, da "v tem drugem tednu cepljenja v Franciji niso opazili resnih škodljivih učinkov" .
Premajhna deklaracija
V resnici bi lahko zlasti v Franciji - ne da bi si nasprotovali - rekli, da so bili vsi možni resni škodljivi učinki izčrpno prijavljeni (prijavljeni) 31 regionalnim centrom za farmakovigilanco (CRPV), ki so razpršeni po celotnem območju. nacionalno ozemlje?
Ovire
Iz izkušenj lahko trdimo, da farmakovigilanca trpi zaradi premajhnega poročanja. Še huje, ugotovljene so ovire pri izjavi (glej članek CTIAP z dne 21. septembra 2019 z naslovom "PRAVICA ODZIVA NA OUEST-FRANCIJA." Upravni prepir "o zdravstveni budnosti. Nadaljevanje" Ovire za farmakovigilanco ": naše opozorilo je posredoval ANSM " ) .
"Organizirana nekaznovanost"
Poleg tega je v začetku julija 2018 neodvisni pregled Prescrire objavil članek z naslovom »Farmacevtska podjetja: organizirana nekaznovanost« . Ta članek nas obvešča, da je "inšpekcijski pregled, opravljen leta 2012", pokazal, da podjetje "ni analiziralo ali posredovalo agencijam za zdravila več kot 80.000 primerov suma škodljivih učinkov na 19 zdravil" . Za ko je sprejela "ponižno in skesano odnos" , je podjetje "iskanih uspešno" pridobitev "o prizanesljivosti evropskih organov" . Pregon se zato "ustavi" . Podjetje"Ne bo treba plačati skoraj 700 milijonov dolarjev globe . "
Tiskanje je razkrilo signale škodljivih učinkov v tujini, ki še niso potrjeni
Zdi se, da teh domnevnih škodljivih učinkov ni prenašal ves francoski tisk.
Glej članek z dne 6. januarja 2021 (točka "IV-C").
II.5- Druge negotovosti glede razmerja med koristjo in tveganjem
Podatki, ki jih lahko štejemo za relativno zadostne, se nanašajo na ljudi, stare od 18 do 75 let, učinkovitost pri blagih do zmernih oblikah, pa tudi na profil kratkotrajnih neželenih učinkov.
Vendar pa se MA izda od starosti 16 let.
Cepljenje se začne pri ljudeh, starejših od 75 let, ki so bili med kliničnimi preskušanji (pri ljudeh) slabo zastopani.
Prav tako razmerje med koristjo in tveganjem tega cepiva pri nosečnicah ni znano. Vendar se zdi, da pozornost mladih žensk v rodni dobi ni dovolj pritegnjena: vprašanje kontracepcije ni obravnavano.
Negotovosti zadevajo tudi doječe ženske, imunsko oslabljene, ljudi, ki so že imeli Covid-19, ljudi z rakom, ljudi z odpovedjo ledvic.
Razmerje med koristjo in tveganjem ni znano v primeru mutacij Sars-CoV-2.
Učinkovitost tega cepiva pri prenosu virusa še ni znana. Zaenkrat je treba še naprej spoštovati zlasti kretnje pregrade.
Učinkovitost in tveganja na srednji in dolgi rok niso znana. Zdi se, da bi bil tekoči projekt namenjen cepljenju ljudi v skupini, ki je prejemala placebo; zaradi česar bi bila analiza podatkov vsaj otežena: primerjava učinkovitosti in škodljivih učinkov med cepljeno skupino in kontrolno skupino (placebo) bo zapletena ali celo nemogoča.
(Za podrobnejšo analizo glej zlasti konferenco 17. decembra 2020).
Vmesni zaključek (iz poglavja "II")
Taka motnja je v nasprotju z besedami ministra za solidarnost in zdravje, ki to podpira v "PORTFOLIO" : "Če bodo zdravstveni delavci poklicani, da prevzamejo vodilno vlogo v kampanji cepljenja, ne bodo prepuščeni sami sebi. Dolžni smo vam, dolgujem vam jasne informacije in absolutno preglednost. To je namen dokumentov, ki so vam jih poslali. "
Opomba: cepivo in "zvezdniki" (zlasti s televizije)
Zaradi pomanjkanja zadostnih in preverljivih dokazov o odprtih vprašanjih v zvezi z razmerjem med koristjo in tveganjem tega cepiva ukrepajo medicinski, farmacevtski, novinarski, politični »zvezdniki« . Počutili bi se vloženi v civilizacijsko poslanstvo do sodržavljanov (preostalega prebivalstva, ki šteje več milijonov ljudi), ki bi bili nevedni.
Pred kamerami so "dali zgled", pravijo.
Bi vzeli ljudi za "otroke" ?
Zdi se, da nista razumela, da "slavnih" ne smemo zamenjati z razvpitostjo.
Zdi se, da vedno prezrejo, da "slavna oseba" ni del meril za ocenjevanje razmerja med koristjo in tveganjem zdravila (cepiva).
Kakšen žalosten "spektakel" ...
In na srečo gledalcev, da je omenjeno cepivo v obliki injekcije; in ne v rektalni obliki (pršilo, svečke itd.) ...
III- Sklep
Zaradi teh neizčrpnih razlogov, navedenih zgoraj, bi lahko to cepivo, vsaj uporabo tega zdravila in razširjenih informacij o njegovem razmerju med koristjo in tveganjem, civilno sodišče opredelilo kot "pomanjkljivo" . .
Kar zadeva kazensko odgovornost, bi to že močno vplivalo zlasti na zdravstvene delavce, za katere se zdi, da igrajo vlogo pri podpori nekaterih govorov, ki javno spodbujajo neuravnotežene, zavajajoče informacije, ki so v nasprotju s podatki, ki jih je pridobila znanost. Nekateri si celo upajo odkrito zagovarjati odpravo korakov, potrebnih za pridobitev privolitve (temeljna svoboda, zaščitena z notranjim in zunanjim pravom). Drugi si celo upajo žaliti ljudi, ki se upravičeno sprašujejo o razmerju med koristjo in tveganjem tega novega cepiva; in čeprav je samo MA "Pogojno" : to pomeni, da se ta MA za eno leto podeli zdravilu (cepivu), katerega razmerje med koristjo in tveganjem je premalo znano in ki ga je treba potrditi s pripravo dodatnih študij.
Poleg tega je v "listu: FOKUSIRANJE ODGOVORNOSTI" omenjenega "PORTFOLIO", ki ga je ustanovilo Ministrstvo za solidarnost in zdravje, jasno opozorjeno, da je "popolno nadomestilo zdravstvenih nesreč, ki jih je mogoče pripisati negovalnim dejavnostim, ki se izvajajo ob kampanjo cepiva proti covid-19 bo v okviru nacionalne solidarnosti zagotovil ONIAM [Nacionalni urad za odškodnine za zdravstvene nesreče] (...) Ta podpora nacionalne solidarnosti pa zdravstvenih delavcev ne izvzema iz odgovornosti (...) « .
Zgodovina medicine kljub temu spominja na okoliščine, ki so povzročile zlasti Nürnberški zakonik .
Odločitev pa je odvisna od suverenega spoštovanja sodnika.
Se nadaljuje…
Drugi odčitki (niso izčrpni)
"TRIBINA. Kar je zavrnjeno profesorju Didierju Raoultu, je dovoljeno drugim «( LE POINT , 7. junij 2020)
"Covid-19 in hidroksiklorokin:" empirizem ", zavrnjen profesorju Didierju Raoultu (v nujnih primerih), vendar tolerirajo" pediatrična zdravila "(v sedanji praksi)" ( CTIAP , 19. september 2020)
https://ctiapchcholet.blogspot.com/2021/01/le-vaccin-contre-la-covid-19.html
*
https://www.brighteon.com/ffaf92a3-7317-4cdc-bead-121a7ed5cf36

*
Rezultati študije o informiranem soglasju v zvezi z eksperimentalnim cepivom: COVID-19 cepiva, zasnovana tako, da izzovejo nevtralizacijska protitelesa, lahko prejemnike cepiva senzibilizirajo za hujšo bolezen, kot če ne bi bili cepljeni. Cepiva za SARS, MERS in RSV še nikoli niso bila odobrena, podatki, pridobljeni pri razvoju in testiranju teh cepiv, pa kažejo na resno mehanistično skrb: da lahko cepiva, zasnovana empirično z uporabo tradicionalnega pristopa (sestavljena iz nemodificiranega ali minimalno modificiranega koronavirusnega virusa, ki izzove nevtralizacijska protitelesa), naj bodo sestavljena iz beljakovin, virusnega vektorja, DNA ali RNA in ne glede na način dostave, poslabšajo bolezen COVID-19 zaradi od protiteles odvisnega povečanja (ADE). To tveganje je v protokolih kliničnih preskušanj in obrazcih soglasja za preskušanja cepiva COVID-19, ki so v teku, dovolj prikrito, da je malo verjetno, da bi ga bolniki ustrezno razumeli, kar onemogoča resnično informirano privolitev udeležencev v teh preskušanjih.
Zaključki študije in klinične posledice: Posebno in pomembno tveganje COVID-19 za ADE bi moralo biti in bi moralo biti vidno in neodvisno razkrito preiskovanim osebam, ki so trenutno v poskusih cepiva, pa tudi tistim, ki se nabirajo za poskuse, in prihodnjim bolnikom po odobritvi cepiva, da se izpolnijo standardi medicinske etike o razumevanju bolnikov za informirano privolitev.
https://pubmed.ncbi.nlm.nih.gov/33113270/
*
Zdi se, da želi vlada opustiti vso previdnost in hiteti s podaljšanjem potnega lista za cepivo , zaradi česar bo deloval kot nekakšno dovoljenje s točkami. Toda to bi bila napaka, saj bi njegove ukrepe izpodbijali na italijanskem sodišču, sodišču in morda tudi evropskem sodišču za človekove pravice, pojasnjuje Alessandro Mangia, redni profesor ustavnega prava na katoliški univerzi v Milanu. »Če bom korak za korakom še naprej razširjal seznam omejitev do pravice do kroženja ali do srečanja, če nimam dovoljenja, na koncu prikrito uvedem ne obveznost, ampak pogoj za cepljenje. In na koncu je to ukrep, enakovreden obveznosti. "
Ali to pomeni, da gre za poskusno cepivo, kot mnogi pravijo, in kot je rekel Figliuolo?
Eksperimentalno cepivo je nenatančen izraz. Izvedene so bile vse faze kliničnih preskušanj, ki jih predvideva evropska zakonodaja. In evropska zakonodaja je veliko strožja od tiste, ki je v ZDA zaupana FDA. Gre za to, da so bile faze izvedene vzporedno - torej istočasno - in ne zaporedno, po fazah, kot je običajno. In očitno je: glede na situacijo je bilo treba pospešiti. Na cepljenje niso mogli čakati 10-15 let.
To tudi vemo. Kakšen je vaš ugovor, profesor?
To je predstavljeno kot birokratska racionalizacija. Toda v resnici ta postopek onemogoča oceno srednjeročnih in dolgoročnih tveganj, značilnih za vsako zdravilo ali cepivo, dano v obtok. Zato njegova izdaja ne s standardnim dovoljenjem, da ostane v jeziku reg. 726/2004, ampak s pogojnim dovoljenjem. Skratka, dosedanja tehnična ocena je povzetek in začasna ocena, ki ji lahko poteče v enem letu po objavi. Z možnostjo podaljšanja. Če bi bili v normalnih razmerah, bi imeli standardno dovoljenje EMA, in vse to ne bi imelo smisla. To je bistvo vsega.
Za kateri razlog?
Ker če bi šlo zgolj za cepljenje proti tuberkulozi ali hepatitisu, teh težav ne bi bilo. Ne gre za to, da so ta cepiva eksperimentalna, karkoli je Figliuolo pomotoma rekel ali kako se ponavlja. Za razliko od cepiv proti črnim kozam ali proti tuberkulozi ta cepiva niso testirana in dokončno odobrena. Pošiljajo se z začasnim dovoljenjem, ki se pregleda vsakih 12 mesecev. Jasneje od tega je le spletno mesto EMA.
In tukaj se, če dovolite, prikrade občutek, da se morate spoprijeti s povsem političnim problemom.
Žal še nismo izumili časovnega stroja, da bi vedeli, kakšni bodo učinki teh cepiv na srednji in dolgi rok. Zato upravičeno pogojno dovoljenje. Karkoli ljudje rečejo, je EMA precej resna stvar. V igri so ogromne odgovornosti, predvsem kaznive. In kljub predstavljivim pritiskom so njeni uradniki podvrženi.
Še vedno se ponavlja, da "nihče ne more biti prisiljen v posebno zdravstveno obravnavo, razen po zakonu" (32 Konst.) In hkrati se nasprotja stigmatizirajo z navajanjem "interesa skupnosti", ki ga ne bi upoštevali upoštevanje. Kako se odzove?
Odgovorim tako, da se spomnim dveh stvari. Po mnenju ustavnega sodišča so zakonodajalca od leta 2002 zavezali rezultati tehničnih ocen zdravstvenih zadev. Se pravi iz rezultatov poskusov. Sodba Onida in odločba Cartabia iz leta 2018. Preizkusi, ki v tem primeru zagotavljajo povzetek in v vsakem primeru ne dokončno oceno. Povzetki preiskav in začasni, izvedeni v imenu nujnosti, niso podlaga za uvedbo zahteve po cepljenju, tudi za omejene kategorije.
In druga stvar, ki si jo morate zapomniti?
To je tisto umetniško delo. 32, ki jih vsi pozabijo omeniti. In to je tisto, kar pravi, da tudi če deluje po zakonu, zakonodajalec "v nobenem primeru ne more kršiti meja, ki jih postavlja spoštovanje človeške osebe". Čudno, da ga nihče več ne omenja.
Ali ni to malo generično? Kaj to pomeni?
Vidite, ne gre za to, da v ustanovni skupščini niso bili pripravljeni, niti za to, da se ti problemi takrat niso pojavili. Tako zelo, da se je 28. januarja 1947 član skupščine po imenu Aldo Moro predstavil Komisiji in pojasnil, da so se zdravniki skupščine obrnili nanj in ga prosili, naj uvede omejitve pooblastil zakonodajalca za urejanje obveznih zdravljenj. Moro nam pravi, da je bil to "problem sterilizacije in druge pomožne težave".
Sterilizacija, ste rekli?
Seveda. Leta 1947 se je nedavnih spomin na nekatera dejstva. Skratka, Morovemu predlogu nasprotujejo različne volivke in volivci, med drugim tudi tisti Umberto Nobile, ki je po tem, ko je bil v rdečem šotoru pri Polu z Regio Aeronautica, izvoljen za prvega namestnika PCI takoj za Togliattijem. Nobile podpira možnost sterilizacije, ker naj bi zakon preprečil, da bi na svet prišli nesrečniki, ki so namenjeni dednim boleznim.
In kako se konča?
Konča se z glasovanjem, v ustavi pa je zapisano, da zakonodajalec "ne more kršiti meja spoštovanja človeške osebe". To ni splošno pravilo. Kot je Moro sam v Komisiji dobro poudaril, je ovira pravilo, "da se izognemo temu, da zakon zaradi splošnih premislekov in slabo razumljenega varstva kolektivnih interesov" predvideva takšno obravnavo.
*
For the US: Read the FDA approval letter please. This is how they scam you, by making it nearly impenetrable. Below, I identify the two most relevant passages. The rest of the letter is repetitive blather to keep your head spinning. https://www.fda.gov/media/150386/download
page 2 last line, footnote: here FDA quietly admits that the licened Pfizer vaccine and the authorized Pfizer vaccine are identical w.r.t. safety and efficacy, but they are "legally distinct." That's code for one has manufacturer liability, while the other doesn't. It is also code for "we don't want to impose a mandate on the EUA product 'cause it is illegal, but we can probably get away with a mandate on the licensed product."
page 12 AA. This tells you that yes, we licensed the vaccine, but...there is a lot of the old vaccine out there, actually "a significant amount" and this amount will be considered an EUA vaccine and will continue to be used. Maybe for a very long time.
Now, why would they do that? Why specify that identical versions of the product will be legally different? Because they need the license to impose the mandates. But they need the EUA to evade liability.
Along with the license comes liability for the manufacturer. (But all EUA products were given a liability shield.)
Unfortunately, our federal overlords would prefer us to be without recourse if we are injured, rather than have Pfizer risk defending its product in court.
So, my inference is that the feds want the public to THINK the vaccine they are receiving is licensed, which will make people submit because they believe it can now be mandated, but instead the public is almost certain to receive the EUA vials instead, to save Pfizer's behind.
* You will be able to tell the difference when you see the vaccine vial. The letter explains that the COMIRNATY-labelled vials will be the licensed ones, and the others (under EUA) will say something like Pfizer BioNTech Covid-19 Vaccine without the brand name COMIRNATY.
Yes, a stingy CICP injury program exists, which I have previously written about, but it has not paid out for a single Covid vaccine injury, last time I checked.
How did I figure this out? Because I have read other FDA approval letters, and this one had significant weasel wording. And I have seen other FDA tricks. So it just took me awhile to identify what was being hidden. I could be wrong. But then we would need another explanation for the language in the approval letter. I'm happy to entertain other interpretations. --meryl
P.S. Pfizer made $33 billion so far this year on its mRNA vaccine.
*
Does the FDA think these data justify the first full approval of a covid-19 vaccine?
The FDA should demand adequate, controlled studies with long term follow up, and make data publicly available, before granting full approval to covid-19 vaccines, says Peter Doshi
On 28 July 2021, Pfizer and BioNTech posted updated results for their ongoing phase 3 covid-19 vaccine trial. The preprint came almost a year to the day after the historical trial commenced, and nearly four months since the companies announced vaccine efficacy estimates “up to six months.”
But you won’t find 10 month follow-up data here. While the preprint is new, the results it contains aren’t particularly up to date. In fact, the paper is based on the same data cut-off date (13 March 2021) as the 1 April press release, and its topline efficacy result is identical: 91.3% (95% CI 89.0 to 93.2) vaccine efficacy against symptomatic covid-19 through “up to six months of follow-up.”
The 20 page preprint matters because it represents the most detailed public account of the pivotal trial data Pfizer submitted in pursuit of the world’s first “full approval” of a coronavirus vaccine from the Food and Drug Administration. It deserves careful scrutiny.
The elephant named “waning immunity”
Since late last year, we’ve heard that Pfizer and Moderna’s vaccines are “95% effective” with even greater efficacy against severe disease (“100% effective,” Moderna said).
Whatever one thinks about the “95% effective” claims (my thoughts are here), even the most enthusiastic commentators have acknowledged that measuring vaccine efficacy two months after dosing says little about just how long vaccine-induced immunity will last. “We’re going to be looking very intently at the durability of protection,” Pfizer senior vice president William Gruber, an author on the recent preprint, told the FDA’s advisory committee last December.
The concern, of course, was decreased efficacy over time. “Waning immunity” is a known problem for influenza vaccines, with some studies showing near zero effectiveness after just three months, meaning a vaccine taken early may ultimately provide no protection by the time “flu season” arrives some months later. If vaccine efficacy wanes over time, the crucial question becomes what level of effectiveness will the vaccine provide when a person is actually exposed to the virus? Unlike covid vaccines, influenza vaccine performance has always been judged over a full season, not a couple months.
And so the recent reports from Israel’s Ministry of Health caught my eye. In early July, they reported that efficacy against infection and symptomatic disease “fell to 64%.” By late July it had fallen to 39% where Delta is the dominant strain. This is very low. For context, the FDA’s expectation is of “at least 50%” efficacy for any approvable vaccine.
Now Israel, which almost exclusively used Pfizer vaccine, has begun administering a third “booster” dose to all adults over 40. And starting 20 September 2021, the US plans to follow suit for all “fully vaccinated” adults eight months past their second dose.
Delta may not be responsible
Enter Pfizer’s preprint. As an RCT reporting “up to six months of follow-up,” it is notable that evidence of waning immunity was already visible in the data by the 13 March 2021 data cut-off.
“From its peak post-dose 2,” the study authors write, “observed VE [vaccine efficacy] declined.” From 96% to 90% (from two months to
But although this additional information was available to Pfizer in April, it was not published until the end of July.
And it’s hard to imagine how the Delta variant could play a real role here, for 77% of trial participants were from the United States, where Delta was not established until months after data cut-off.
Waning efficacy has the potential to be far more than a minor inconvenience; it can dramatically change the risk-benefit calculus. And whatever its cause—intrinsic properties of the vaccine, the circulation of new variants, some combination of the two, or something else—the bottom line is that vaccines need to be effective.
Until new clinical trials demonstrate that boosters increase efficacy above 50%, without increasing serious adverse events, it is unclear whether the 2-dose series would even meet the FDA’s approval standard at six or nine months.
The “six month” preprint based on the 7% of trial participants who remained blinded at six months
The final efficacy timepoint reported in Pfizer’s preprint is “from four months to the data cut-off.” The confidence interval here is wider than earlier time points because only half of trial participants (53%) made it to the four month mark, and mean follow-up is around 4.4 months (see footnote).
This all happened because starting last December, Pfizer allowed all trial participants to be formally unblinded, and placebo recipients to get vaccinated. By 13 March 2021 (data cut-off), 93% of trial participants (41,128 of 44,060; Fig 1) were unblinded, officially entering “open-label followup.” (Ditto for Moderna: by mid April, 98% of placebo recipients had been vaccinated.)
Despite the reference to “six month safety and efficacy” in the preprint’s title, the paper only reports on vaccine efficacy “up to six months,” but not from six months. This is not semantics, as it turns out only 7% of trial participants actually reached six months of blinded follow-up (“8% of BNT162b2 recipients and 6% of placebo recipients had ≥6 months follow-up post-dose 2.”) So despite this preprint appearing a year after the trial began, it provides no data on vaccine efficacy past six months, which is the period Israel says vaccine efficacy has dropped to 39%.
It is hard to imagine that the <10% of trial participants who remained blinded at six months (which presumably further dwindled after 13 March 2021) could constitute a reliable or valid sample to produce further findings. And the preprint does not report any demographic comparisons to justify future analyses.
Severe disease
With the US awash in news about rising cases of the Delta variant, including among the “fully vaccinated,” the vaccine’s efficacy profile is in question. But some medical commentators are delivering an upbeat message. Former FDA commissioner Scott Gottlieb, who is on Pfizer’s board, said: “Remember, the original premise behind these vaccines were [sic] that they would substantially reduce the risk of death and severe disease and hospitalization. And that was the data that came out of the initial clinical trials.”
Yet, the trials were not designed to study severe disease. In the data that supported Pfizer’s EUA, the company itself characterized the “severe covid-19” endpoint results as “preliminary evidence.” Hospital admission numbers were not reported, and zero covid-19 deaths occurred.
In the preprint, high efficacy against “severe covid-19” is reported based on all follow-up time (one event in the vaccinated group vs 30 in placebo), but the number of hospital admissions is not reported so we don’t know which, if any, of these patients were ill enough to require hospital treatment. (In Moderna’s trial, data last year showed that 21 of 30 “severe covid-19” cases were not admitted to hospital; Table S14).
And on preventing death from covid-19, there are too few data to draw conclusions—a total of three covid-19 related deaths (one on vaccine, two on placebo). There were 29 total deaths during blinded follow-up (15 in the vaccine arm; 14 in placebo).
The crucial question, however, is whether the waning efficacy seen in the primary endpoint data also applies to the vaccine’s efficacy against severe disease. Unfortunately, Pfizer’s new preprint does not report the results in a way that allows for evaluating this question.
Approval imminent without data transparency, or even an advisory committee meeting?
Last December, with limited data, the FDA granted Pfizer’s vaccine an EUA, enabling access to all Americans who wanted one. It sent a clear message that the FDA could both address the enormous demand for vaccines without compromising on the science. A “full approval” could remain a high bar.
But here we are, with FDA reportedly on the verge of granting a marketing license 13 months into the still ongoing, two year pivotal trial, with no reported data past 13 March 2021, unclear efficacy after six months due to unblinding, evidence of waning protection irrespective of the Delta variant, and limited reporting of safety data. (The preprint reports “decreased appetite, lethargy, asthenia, malaise, night sweats, and hyperhidrosis were new adverse events attributable to BNT162b2 not previously identified in earlier reports,” but provides no data tables showing the frequency of these, or other, adverse events.)
It’s not helping matters that FDA now says it won’t convene its advisory committee to discuss the data ahead of approving Pfizer’s vaccine. (Last August, to address vaccine hesitancy, the agency had “committed to use an advisory committee composed of independent experts to ensure deliberations about authorization or licensure are transparent for the public.”)
Prior to the preprint, my view, along with a group of around 30 clinicians, scientists, and patient advocates, was that there were simply too many open questions about all covid-19 vaccines to support approving any this year. The preprint has, unfortunately, addressed very few of those open questions, and has raised some new ones.
I reiterate our call: “slow down and get the science right—there is no legitimate reason to hurry to grant a license to a coronavirus vaccine.”
FDA should be demanding that the companies complete the two year follow-up, as originally planned (even without a placebo group, much can still be learned about safety). They should demand adequate, controlled studies using patient outcomes in the now substantial population of people who have recovered from covid. And regulators should bolster public trust by helping ensure that everyone can access the underlying data.
Peter Doshi, senior editor, The BMJ.
Competing interests: I helped organize the Coalition Advocating for Adequately Licensed Medicines (CAALM), which has formally petitioned the FDA to refrain from fully approving any covid-19 vaccine this year (docket FDA-2021-P-0786). A full list of competing interests is available here.
Provenance: commissioned; externally peer-reviewed.
Footnote: Calculations in this article are as follows. “About 1 month” past month 4 is based on the final row of Fig 2 in the preprint: 1030/12670*12 = 0.98 months (vaccine group) and 895/11802*12 = 0.91 months (placebo group). “53%” is based on Fig 2: (12670+11802)/(23040+23037). “4.4 months” is based on the average of 8412/22505*12 = 4.5 (vaccine) and 8124/22434*12 = 4.3 (placebo) in Fig 2.
*
Buried in the fine print of Monday’s approval by the U.S. Food and Drug Administration of the Pfizer Comirnaty COVID vaccine are two critical facts that affect whether the vaccine can be mandated, and whether Pfizer can be held liable for injuries.
Monday, the U.S. Food and Drug Administration (FDA) approved a biologics license application for the Pfizer Comirnaty vaccine.
The press reported that vaccine mandates are now legal for military, healthcare workers, college students and employees in many industries. New York City Mayor Bill de Blasio has now required the vaccine for all teachers and school staff. The Pentagon is proceeding with its mandate for all military service members.
But there are several bizarre aspects to the FDA approval that will prove confusing to those not familiar with the pervasiveness of the FDA’s regulatory capture, or the depths of the agency’s cynicism.
First, the FDA acknowledges that while Pfizer has insufficient stocks of the newly licensed Comirnaty vaccine available, there is “a significant amount” of the Pfizer-BioNTech COVID vaccine — produced under Emergency Use Authorization (EUA) — available for use.
The FDA decrees that the Pfizer-BioNTech vaccine under the EUA should remain unlicensed but can be used “interchangeably” (page 2, footnote 8) with the newly licensed Comirnaty product.
Second, the FDA pointed out that both the licensed Pfizer Comirnaty vaccine, and the existing vaccine are “legally distinct,” but proclaims that their differences do not “impact safety or effectiveness.”
There is a huge real-world difference between products under an EUA compared with those that FDA has fully licensed. EUA products are experimental under U.S. law.
Both the Nuremberg Code and federal regulations provide that no one can force a human being to participate in this experiment. Under 21 U.S. Code Sec.360bbb-3(e)(1)(A)(ii)(III), “authorization for medical products for use in emergencies,” it is unlawful to deny someone a job or an education because they refuse to be an experimental subject. Instead, potential recipients have an absolute right to refuse EUA vaccines.
U.S. laws, however, permit employers and schools to require students and workers to take licensed vaccines.
EUA-licensed vaccines have an extraordinary liability shield under the 2005 Public Readiness and Preparedness Act. Vaccine manufacturers, distributors, providers and government planners are immune from liability. The only way an injured party can sue is if he or she can prove willful misconduct, and if the U.S. government has also brought an enforcement action against the party for willful misconduct. No such lawsuit has ever succeeded.
*
Does the FDA Think These Data Justify the First Full Approval of a COVID-19 Vaccine?
The U.S. Food and Drug Administration should have demanded adequate, controlled studies with long-term follow up, and made data publicly available, before granting full approval to COVID-19 vaccines, says BMJ Associate editor Peter Doshi.
*
On Tuesday, Dr. Robert Malone, the inventor of mRNA vaccine technology, appeared on Bannon’s War Room to explain the Covid vaccine’s FDA “approval status.”
According to Dr. Malone, the Pfizer vaccine looks to be incompletely approved, and they were only granted an extension under the Emergency Use Authorization (EUA), which provides them with full liability protection.
Join The True Defender Telegram Chanel Here: https://t.me/TheTrueDefender
The FDA has approved a Biologics License Application (BLA) for the Comirnaty vaccine, which is “substantially similar but not necessarily identical” and has not yet been developed.
“It’s [Comirnaty] not available at all. So, the little trick they’ve pulled here is that they’ve sent out two separate letters.
On Tuesday, Dr. Robert Malone, the inventor of mRNA vaccine technology, appeared on Bannon’s War Room to explain the Covid vaccine’s FDA “approval status.”
According to Dr. Malone, the Pfizer vaccine looks to be incompletely approved, and they were only granted an extension under the Emergency Use Authorization (EUA), which provides them with full liability protection.
The FDA has approved a Biologics License Application (BLA) for the Comirnaty vaccine, which is “substantially similar but not necessarily identical” and has not yet been developed.
“It’s [Comirnaty] not available at all. So, the little trick they’ve pulled here is that they’ve sent out two separate letters.
The FDA put out two letters about the two vaccines, confusing the public and participating in the manipulation of the fake-news media.
Rather than awarding full approval to both, Pfizer is extending the EUA so that Pfizer’s vaccine can be administered when Comirnaty vaccine supply is “restricted or non-existent.”
*
Two important facts about EUA drugs and vaccines
We know there are some important differences between EUA drugs and vaccines, and fully licensed drugs or vaccines.
We also know these two facts about EUA products:
- EUA vaccines are designated as experimental or investigational products under U.S. law. As such, they cannot be mandated. You have the right to refuse, without suffering consequences.
- EUA vaccines have a huge liability shield that protects everyone involved with the product from being sued. If you are injured by an EUA vaccine, the only way to obtain compensation for damages is to apply to the Countermeasures Injury Compensation Program (CICP), which might cover unpaid medical expenses and lost wages only. However, only 3% of claims made have been compensated, and so far the program has approved no claims for COVID vaccine injuries.
Some say the CICP, which is run through the U.S. Department of Health and Human Service and does not give petitioners the right to a judge or jury, withholds due process from injured Americans.
Yet this is the only pathway by which an injured party can seek help after receiving an EUA vaccine or drug.
It’s right there in the fact sheet — FDA says it’s ‘your choice’
Legally, in order to mandate a vaccine, the vaccine must be fully approved. However, once a vaccine for use in adults moves from an EUA product to a licensed everyday product, it loses its liability shield.
We believe it is likely the FDA was instructed to find a way to both license the Pfizer vaccine — so mandates would be legally supported — while also retaining the vaccine’s liability shield.
The FDA could not find a way to do this under existing law. So instead, as we reported Aug. 24 in The Defender, the agency chose to create confusion regarding the legal status of the two Pfizer-BioNTech vaccines. In a document we discuss here for the first time — the fact sheet required to be given to recipients of either the Comirnaty or Pfizer-BioNTech COVID vaccine — the FDA acknowledges the facts we have just presented.
But the FDA also has added something new — the final sentence of the fact sheet states:
“This EUA for the Pfizer-BioNTech COVID-19 Vaccine and COMIRNATY will end when the Secretary of HHS determines that the circumstances justifying the EUA no longer exist or when there is a change in the approval status of the product such that an EUA is no longer needed.”
According to this fact sheet, the FDA also has designated the licensed Comirnaty vaccine as an EUA product. By doing so, the FDA has guaranteed the Comirnaty vaccine the same liability shield as the EUA Pfizer-BioNTech vaccine.
However, that means the Comirnaty vaccine cannot be mandated. The FDA admits this in the fact sheet, where it states:
“WHAT IF I DECIDE NOT TO GET COMIRNATY (COVID-19 VACCINE, mRNA) OR THE PFIZER-BIONTECH COVID-19 VACCINE?
“Under the EUA, it is your choice to receive or not receive the vaccine. Should you decide not to receive it, it will not change your standard medical care.”
The fact sheet is FDA’s admission, buried in the fine print, that no one can currently be mandated to receive any COVID vaccine in the U.S., as all remain under the EUA.
This fact sheet for vaccine recipients is key to avoid being forced to accept an experimental vaccine.
We suggest you print it out, highlight the relevant passages and present it to anyone who tries to force vaccinations on employees.
https://childrenshealthdefense.org/defender/no-one-can-force-you-pfizers-comirnaty-vaccine/?utm_source=salsa&eType=EmailBlastContent&eId=4050fe4d-20c9-40cb-bba6-a999ea641ee3&fbclid=IwAR1Iiiua2h6y9yz3DoJdYpYCARYSou2grMUw2nacQ2wnOR1iP7KbufAngjo
https://www.fda.gov/media/144414/download
Prevod: Meryl Nass, dr. med.: Dve pomembni dejstvi o zdravilih in cepivih z dovoljenjem za uporabo v sili (EUA)
*

*
Prevara FDA pri odobritvi Pfizer cepiva: https://principia-scientific.com/major-law-firm-confirms-fda-deceived-america-with-its-approval-of-pfizer-vax
*
Odvetnik Domen Gorenc: V kolikor je bilo namreč res, da je bilo s cepivom Janssen cepljenih več kot dvesto mladoletnih otrok, smo bili v Republiki Sloveniji s pravnega vidika, po mojem mnenju, v 21. stoletju priča izvajanju poskusov na človeku. Navedeno je seveda izrecno prepovedano tako z 18. členom Ustave RS, Kazenskim zakonikom (KZ-1), Zakonom o pacientovih pravicah, Zakonom o zdravniški službi, Zakonom o zdravilih, Zakonom o zdravstveni dejavnosti, kot seveda tudi s Kodeksom zdravniške etike. Med mednarodnopravnimi akti velja, poleg že večkrat citirane Evropske konvencije o človekovih pravicah in Nürnberškega kodeksa, ki določa absolutno nujnost privolitve v medicinski poskus, na tem mestu izpostaviti predvsem Helsinško deklaracijo in s strani Sveta Evrope izdano Oviedsko konvencijo s protokoli o človekovih pravicah v zvezi z biomedicino.
*
Znanstveniki tožijo FDA za podatke, na osnovi katerih so odobrili Pfizer cepivo:
May 30, 2021
